

Overview of Scarf#
Jump to installation | Try live code | Source code on Github
What is Scarf#
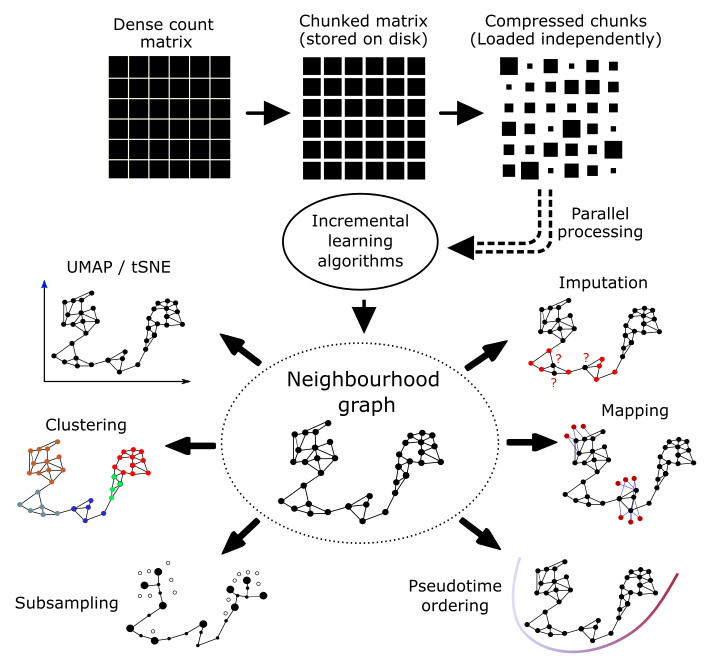
Scarf is a Python package that performs memory-efficient analysis of single-cell genomics data. Using an efficient data chunking process (using Zarr and Dask) Scarf manages to perform the core steps of single-cell genomics data analysis with very low memory consumption. Scarf’s core step is to efficiently generate a neighbourhood graph (KNN graph) of cells. This graph forms the basis for downstream steps of the analysis thus maximizing concordance between those steps. Read below to see what cool new features Scarf has on offer.
Scarf is published!
The article describing Scarf is published in Nature Communications and is available here
What does Scarf offer#
Analyze atlas scale scRNA-Seq datasets on your laptop (up to 4 million cells tested)
Perform analysis of scATAC-Seq datasets (datasets with up to 700K cells and 1M peaks tested)
Parallel implementations of UMAP and tSNE (SG-tSNE) for quick cell embedding
Perform hierarchical clustering that gives interpretable cluster relationships
Sub-sample highly representative cells using state-of-the-art TopACeDo method
Perform projections of cells from one dataset onto another or integrate multiple datasets
Why use Scarf#
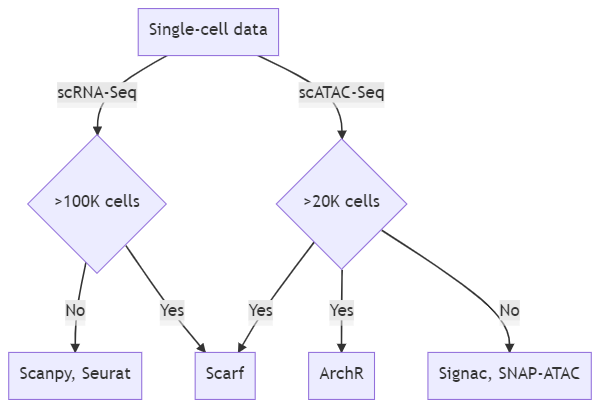
The following flowchart can help one decide which tool to choose with respect to the size of the data. This flowchart assumes that you do not have access or simply do not want to use a system with large RAM capacity. Only a few popular tools have been mentioned here, this is not an exhaustive list.
Example usage scenarios#
You have generated a knockdown model of some stem cells and want to see which lineages are affected as a result of this perturbation. So you perform scRNA-Seq of this perturbed stem cell derived populations. You download an atlas-scale data available for this tissue system but the data is too large and can’t be analyzed on your laptop. Using Scarf you can quickly generate a UMAP of the atlas scale data and visualize all the author annotated clusters. Now you can project your perturbed population over this map to check how the heterogeneity has been affected due the perturbation.
There is an atlas-scale (say more than 500K cells) dataset available for some embryonic tissue. You want to redo the cell trajectory analysis with a new and promising tool that just came out on Bioarxiv. However, the dataset is too large to be analyzed on your laptop. Using Scarf, you can perform clustering on the data on your laptop and run Scarf’s cell downsampling algorithm to select the most representative cells from the clusters of your interest. You can then use this downsampled data and run the trajectory analysis on it.
When not to use Scarf#
There is no reason to not use Scarf, generally. But Scarf currently lacks a lots of functionality that is available elsewhere. The primary reason for this is that we want to guarantee the memory efficiency of those methods.
Scarf development: the future#
The core team is constantly working to improve Scarf’s functionality, add new features and to make the code more robust (aka testing). We aim to extend the Scarf to more single-cell methodologies by including normalization and dimension reduction strategies best suited to those methods.