Workflow for scATAC-Seq data#
If you haven’t already then we highly recommend that you please go the Workflow for scRNA-Seq analysis and also go through this vignette describing data organization and design principles of Scarf.
Here we will go through some of the basic steps of analysis of single-cell ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) data.
%load_ext autotime
import scarf
scarf.__version__
'0.31.4'
time: 814 ms (started: 2025-04-15 15:22:20 +00:00)
1) Fetch and convert data#
We will use 10x Genomics’s singel-cell ATAC-Seq data from peripheral blood mononuclear cells. Like single-cell RNA-Seq, Scarf only needs a count matrix to start the analysis of scATAC-Seq data. We can use fetch_dataset to download the data in 10x’s HDF5 format.
scarf.fetch_dataset(
dataset_name='tenx_10K_pbmc-v1_atacseq',
save_path='scarf_datasets'
)
time: 23.4 s (started: 2025-04-15 15:22:20 +00:00)
The CrH5Reader class provides access to the HDF5 file. We can load the file and quickly check the number of features, and also verify that Scarf identified the assay as an ATAC assay.
reader = scarf.CrH5Reader(
'scarf_datasets/tenx_10K_pbmc-v1_atacseq/data.h5'
)
reader.assayFeats
| ATAC | |
|---|---|
| type | Peaks |
| start | 0 |
| end | 89796 |
| nFeatures | 89796 |
time: 43.3 ms (started: 2025-04-15 15:22:44 +00:00)
writer = scarf.CrToZarr(
reader,
zarr_loc=f'scarf_datasets/tenx_10K_pbmc-v1_atacseq/data.zarr',
chunk_size=(1000, 2000)
)
writer.dump(batch_size=1000)
time: 7.69 s (started: 2025-04-15 15:22:44 +00:00)
2) Create DataStore and filter cells#
We load the Zarr file on using DataStore class. The obtained DataStore object will be our single point on interaction for rest of this analysis. When loaded, Scarf will automatically calculate the number of cells where each peak is present and number of peaks that are accessible in cell (nFeatures). Scarf will also calculate the total number of fragments/cut sites within each cell.
ds = scarf.DataStore(
'scarf_datasets/tenx_10K_pbmc-v1_atacseq/data.zarr',
nthreads=4
)
time: 6.22 s (started: 2025-04-15 15:22:52 +00:00)
We will use auto_filter_cells method which automatically remove outliers from the data. To identify outliers we generate a normal distribution using sample mean and variance. Using this normal distribution Scarf estimates the values with probability less 0.01 (default value) on both ends of the distribution and flags them for removal.
ds.auto_filter_cells()
INFO: 247 cells flagged for filtering out using attribute ATAC_nCounts
INFO: 294 cells flagged for filtering out using attribute ATAC_nFeatures
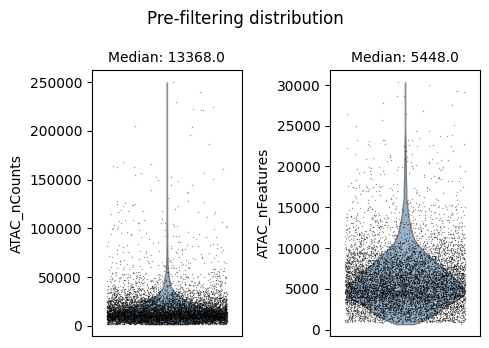
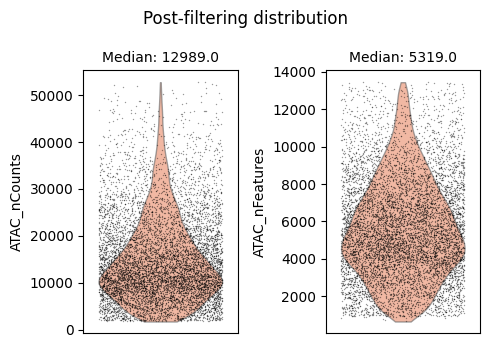
time: 3.59 s (started: 2025-04-15 15:22:58 +00:00)
3) Feature selection#
For scATAC-Seq data, the features are ranked by their TF-IDF normalized values, summed across all cells. The top n features are marked as prevalent_peaks and are used for downstream steps. Here we used top 25000 peaks, which makes for more than quarter of all the peaks which inline with the what has been suggested in other scATAC-Seq analysis protocols.
ds.mark_prevalent_peaks(top_n=25000)
time: 5.37 s (started: 2025-04-15 15:23:01 +00:00)
ds.ATAC.feats.head()
| I | ids | names | I__prevalent_peaks | dropOuts | nCells | stats_I_prevalence | |
|---|---|---|---|---|---|---|---|
| 0 | True | chr1:565107-565550 | chr1:565107-565550 | False | 8645 | 83 | 0.206181 |
| 1 | True | chr1:569174-569639 | chr1:569174-569639 | False | 8529 | 199 | 0.350725 |
| 2 | True | chr1:713460-714823 | chr1:713460-714823 | True | 6242 | 2486 | 1.993573 |
| 3 | True | chr1:752422-753038 | chr1:752422-753038 | True | 8296 | 432 | 0.680701 |
| 4 | True | chr1:762106-763359 | chr1:762106-763359 | True | 6850 | 1878 | 1.570790 |
time: 20.5 ms (started: 2025-04-15 15:23:07 +00:00)
4) KNN graph creation#
For scATAC-Seq datasets, Scarf uses TF-IDF normalization. The normalization is automatically performed during the graph building step. The selected features, marked as prevalent_peaks in feature metadata, are used for graph creation. For the dimension reduction step, LSI (latent semantic indexing) is used rather than PCA. The rest of the steps are same as for scRNA-Seq data.
LSI reduction of scATAC-Seq is known to capture the sequencing depth of cells in the first LSI dimension. Hence, by default, the lsi_skip_first parameter is True but users can override it.
ds.make_graph(
feat_key='prevalent_peaks',
k=21,
dims=50,
n_centroids=100,
lsi_skip_first=True,
)
---------------------------------------------------------------------------
KeyboardInterrupt Traceback (most recent call last)
Cell In[9], line 1
----> 1 ds.make_graph(
2 feat_key='prevalent_peaks',
3 k=21,
4 dims=50,
5 n_centroids=100,
6 lsi_skip_first=True,
7 )
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/scarf/datastore/graph_datastore.py:992, in GraphDataStore.make_graph(self, from_assay, cell_key, feat_key, pca_cell_key, reduction_method, dims, k, ann_metric, ann_efc, ann_ef, ann_m, ann_parallel, rand_state, n_centroids, batch_size, log_transform, renormalize_subset, local_connectivity, bandwidth, update_keys, return_ann_object, custom_loadings, feat_scaling, lsi_skip_first, harmonize, batch_columns, show_elbow_plot, ann_index_fetcher, ann_index_saver)
984 recall = self_query_knn(
985 ann_obj=ann_obj,
986 store=self.zw.create_group(knn_loc, overwrite=True),
987 chunk_size=batch_size,
988 nthreads=self.nthreads,
989 )
990 recall = "%.2f" % recall
--> 992 smoothen_dists(
993 self.zw.create_group(graph_loc, overwrite=True),
994 self.zw[knn_loc]["indices"],
995 self.zw[knn_loc]["distances"],
996 local_connectivity,
997 bandwidth,
998 batch_size,
999 )
1000 if recall is not None:
1001 logger.info(f"ANN recall: {recall}%")
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/scarf/knn_utils.py:132, in smoothen_dists(store, z_idx, z_dist, lc, bw, chunk_size)
128 sigmas, rhos = smooth_knn_dist(
129 kv, k=n_neighbors, local_connectivity=lc, bandwidth=bw
130 )
131 if umap_is_latest:
--> 132 rows, cols, vals, _ = compute_membership_strengths(ki, kv, sigmas, rhos)
133 else:
134 rows, cols, vals = compute_membership_strengths(ki, kv, sigmas, rhos)
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/dispatcher.py:376, in _DispatcherBase._compile_for_args(self, *args, **kws)
374 return_val = None
375 try:
--> 376 return_val = self.compile(tuple(argtypes))
377 except errors.ForceLiteralArg as e:
378 # Received request for compiler re-entry with the list of arguments
379 # indicated by e.requested_args.
380 # First, check if any of these args are already Literal-ized
381 already_lit_pos = [i for i in e.requested_args
382 if isinstance(args[i], types.Literal)]
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/dispatcher.py:904, in Dispatcher.compile(self, sig)
902 with ev.trigger_event("numba:compile", data=ev_details):
903 try:
--> 904 cres = self._compiler.compile(args, return_type)
905 except errors.ForceLiteralArg as e:
906 def folded(args, kws):
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/dispatcher.py:80, in _FunctionCompiler.compile(self, args, return_type)
79 def compile(self, args, return_type):
---> 80 status, retval = self._compile_cached(args, return_type)
81 if status:
82 return retval
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/dispatcher.py:94, in _FunctionCompiler._compile_cached(self, args, return_type)
91 pass
93 try:
---> 94 retval = self._compile_core(args, return_type)
95 except errors.TypingError as e:
96 self._failed_cache[key] = e
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/dispatcher.py:107, in _FunctionCompiler._compile_core(self, args, return_type)
104 flags = self._customize_flags(flags)
106 impl = self._get_implementation(args, {})
--> 107 cres = compiler.compile_extra(self.targetdescr.typing_context,
108 self.targetdescr.target_context,
109 impl,
110 args=args, return_type=return_type,
111 flags=flags, locals=self.locals,
112 pipeline_class=self.pipeline_class)
113 # Check typing error if object mode is used
114 if cres.typing_error is not None and not flags.enable_pyobject:
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/compiler.py:739, in compile_extra(typingctx, targetctx, func, args, return_type, flags, locals, library, pipeline_class)
715 """Compiler entry point
716
717 Parameter
(...) 735 compiler pipeline
736 """
737 pipeline = pipeline_class(typingctx, targetctx, library,
738 args, return_type, flags, locals)
--> 739 return pipeline.compile_extra(func)
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/compiler.py:439, in CompilerBase.compile_extra(self, func)
437 self.state.lifted = ()
438 self.state.lifted_from = None
--> 439 return self._compile_bytecode()
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/compiler.py:505, in CompilerBase._compile_bytecode(self)
501 """
502 Populate and run pipeline for bytecode input
503 """
504 assert self.state.func_ir is None
--> 505 return self._compile_core()
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/compiler.py:473, in CompilerBase._compile_core(self)
471 res = None
472 try:
--> 473 pm.run(self.state)
474 if self.state.cr is not None:
475 break
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/compiler_machinery.py:356, in PassManager.run(self, state)
354 pass_inst = _pass_registry.get(pss).pass_inst
355 if isinstance(pass_inst, CompilerPass):
--> 356 self._runPass(idx, pass_inst, state)
357 else:
358 raise BaseException("Legacy pass in use")
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/compiler_lock.py:35, in _CompilerLock.__call__.<locals>._acquire_compile_lock(*args, **kwargs)
32 @functools.wraps(func)
33 def _acquire_compile_lock(*args, **kwargs):
34 with self:
---> 35 return func(*args, **kwargs)
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/compiler_machinery.py:311, in PassManager._runPass(self, index, pss, internal_state)
309 mutated |= check(pss.run_initialization, internal_state)
310 with SimpleTimer() as pass_time:
--> 311 mutated |= check(pss.run_pass, internal_state)
312 with SimpleTimer() as finalize_time:
313 mutated |= check(pss.run_finalizer, internal_state)
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/compiler_machinery.py:272, in PassManager._runPass.<locals>.check(func, compiler_state)
271 def check(func, compiler_state):
--> 272 mangled = func(compiler_state)
273 if mangled not in (True, False):
274 msg = ("CompilerPass implementations should return True/False. "
275 "CompilerPass with name '%s' did not.")
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/typed_passes.py:112, in BaseTypeInference.run_pass(self, state)
106 """
107 Type inference and legalization
108 """
109 with fallback_context(state, 'Function "%s" failed type inference'
110 % (state.func_id.func_name,)):
111 # Type inference
--> 112 typemap, return_type, calltypes, errs = type_inference_stage(
113 state.typingctx,
114 state.targetctx,
115 state.func_ir,
116 state.args,
117 state.return_type,
118 state.locals,
119 raise_errors=self._raise_errors)
120 state.typemap = typemap
121 # save errors in case of partial typing
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/typed_passes.py:93, in type_inference_stage(typingctx, targetctx, interp, args, return_type, locals, raise_errors)
91 infer.build_constraint()
92 # return errors in case of partial typing
---> 93 errs = infer.propagate(raise_errors=raise_errors)
94 typemap, restype, calltypes = infer.unify(raise_errors=raise_errors)
96 return _TypingResults(typemap, restype, calltypes, errs)
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/typeinfer.py:1066, in TypeInferer.propagate(self, raise_errors)
1063 oldtoken = newtoken
1064 # Errors can appear when the type set is incomplete; only
1065 # raise them when there is no progress anymore.
-> 1066 errors = self.constraints.propagate(self)
1067 newtoken = self.get_state_token()
1068 self.debug.propagate_finished()
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/typeinfer.py:160, in ConstraintNetwork.propagate(self, typeinfer)
157 with typeinfer.warnings.catch_warnings(filename=loc.filename,
158 lineno=loc.line):
159 try:
--> 160 constraint(typeinfer)
161 except ForceLiteralArg as e:
162 errors.append(e)
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/typeinfer.py:692, in IntrinsicCallConstraint.__call__(self, typeinfer)
690 if fnty in utils.OPERATORS_TO_BUILTINS:
691 fnty = typeinfer.resolve_value_type(None, fnty)
--> 692 self.resolve(typeinfer, typeinfer.typevars, fnty=fnty)
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/typeinfer.py:589, in CallConstraint.resolve(self, typeinfer, typevars, fnty)
587 fnty = fnty.instance_type
588 try:
--> 589 sig = typeinfer.resolve_call(fnty, pos_args, kw_args)
590 except ForceLiteralArg as e:
591 # Adjust for bound methods
592 folding_args = ((fnty.this,) + tuple(self.args)
593 if isinstance(fnty, types.BoundFunction)
594 else self.args)
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/typeinfer.py:1560, in TypeInferer.resolve_call(self, fnty, pos_args, kw_args)
1557 return sig
1558 else:
1559 # Normal non-recursive call
-> 1560 return self.context.resolve_function_type(fnty, pos_args, kw_args)
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/typing/context.py:195, in BaseContext.resolve_function_type(self, func, args, kws)
193 # Prefer user definition first
194 try:
--> 195 res = self._resolve_user_function_type(func, args, kws)
196 except errors.TypingError as e:
197 # Capture any typing error
198 last_exception = e
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/typing/context.py:247, in BaseContext._resolve_user_function_type(self, func, args, kws, literals)
243 return self.resolve_function_type(func_type, args, kws)
245 if isinstance(func, types.Callable):
246 # XXX fold this into the __call__ attribute logic?
--> 247 return func.get_call_type(self, args, kws)
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/types/functions.py:308, in BaseFunction.get_call_type(self, context, args, kws)
305 nolitargs = tuple([_unlit_non_poison(a) for a in args])
306 nolitkws = {k: _unlit_non_poison(v)
307 for k, v in kws.items()}
--> 308 sig = temp.apply(nolitargs, nolitkws)
309 except Exception as e:
310 if not isinstance(e, errors.NumbaError):
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/typing/templates.py:490, in ConcreteTemplate.apply(self, args, kws)
488 def apply(self, args, kws):
489 cases = getattr(self, 'cases')
--> 490 return self._select(cases, args, kws)
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/typing/templates.py:284, in FunctionTemplate._select(self, cases, args, kws)
279 def _select(self, cases, args, kws):
280 options = {
281 'unsafe_casting': self.unsafe_casting,
282 'exact_match_required': self.exact_match_required,
283 }
--> 284 selected = self.context.resolve_overload(self.key, cases, args, kws,
285 **options)
286 return selected
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/typing/context.py:635, in BaseContext.resolve_overload(self, key, cases, args, kws, allow_ambiguous, unsafe_casting, exact_match_required)
633 for case in cases:
634 if len(args) == len(case.args):
--> 635 rating = self._rate_arguments(args, case.args, **options)
636 if rating is not None:
637 candidates.append((rating.astuple(), case))
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/typing/context.py:575, in BaseContext._rate_arguments(self, actualargs, formalargs, unsafe_casting, exact_match_required)
573 rate = Rating()
574 for actual, formal in zip(actualargs, formalargs):
--> 575 conv = self.can_convert(actual, formal)
576 if conv is None:
577 return None
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/typing/context.py:551, in BaseContext.can_convert(self, fromty, toty)
547 return Conversion.exact
548 else:
549 # First check with the type manager (some rules are registered
550 # at startup there, see numba.typeconv.rules)
--> 551 conv = self.tm.check_compatible(fromty, toty)
552 if conv is not None:
553 return conv
File ~/checkouts/readthedocs.org/user_builds/scarf/envs/latest/lib/python3.11/site-packages/numba/core/typeconv/typeconv.py:44, in TypeManager.check_compatible(self, fromty, toty)
41 if not isinstance(toty, types.Type):
42 raise ValueError("Specified type '%s' (%s) is not a Numba type" %
43 (toty, type(toty)))
---> 44 name = _typeconv.check_compatible(self._ptr, fromty._code, toty._code)
45 conv = Conversion[name] if name is not None else None
46 assert conv is not Conversion.nil
KeyboardInterrupt:
time: 3min 21s (started: 2025-04-15 15:23:07 +00:00)
5) UMAP reduction and clustering#
Non-linear dimension reduction using UMAP and tSNE are performed in the same way as for scRNA-Seq data. Because, in Scarf the core UMAP step are run directly on the neighbourhood graph, the scATAC-Seq data is handled similar to any other data.
ds.run_umap(
n_epochs=500,
min_dist=0.1,
spread=1,
parallel=True
)
Same goes for clustering as well. The leiden clustering acts on the neighbourhood graph directly.
ds.run_leiden_clustering(resolution=0.6)
Results from both UMAP and Leiden clustering are stored in cell attribute table. Here, because the UMAP and leiden clustering columns have ‘ATAC’ prefixes because they were executed on the default ‘ATAC’ assay. You can also see that the cells that were filtered out (marked by ‘False’ value in column ‘I’) have NaN value for UMAP axes and -1 for clustering
ds.cells.head()
We can visualize the UMAP embedding and the clusters of cells on the embedding
ds.plot_layout(
layout_key='ATAC_UMAP',
color_by='ATAC_leiden_cluster'
)
Those familiar with PBMC datasets might already be able to identify different cell types in the UMAP plot.
6) Calculating gene scores#
The features in snATAC-Seq in form of peak coordinates are hard to interpret by themselves.
Hence, distilling the open chromatin regions in terms of accessible genes can help in identification of cell types.
GeneScores are simply the summation of all the peak fragments that are present within gene bodies and their corresponding promoter region.
Since, marker genes are often better understood than the non-coding regions the GeneScores can be used to annotate cell types.
In Scarf, the users can provide a BED file containing gene annotations. This BED should have no header, should be tab separated and must have the columns in following order:
chromosome identifier
start coordinate
end coordinate
Gene ID
Gene Name
Strand (Optional)
The start/end coordinate can extend through transcription start site (TSS) to include a portion of promoter.
For convenience we have generated such BED files for human and mouse assemblies using the annotation information from GENCODE project. We downloaded the GFF3 format primary chromosome annotations and used Scarf’s GFFReader to convert the files into BED and add promoter offset of 2KB. These BED files containing gene annotations can be downloaded using fetch_dataset command and passing ‘annotations’ parameter.
scarf.fetch_dataset(
dataset_name='annotations',
save_path='scarf_datasets'
)
Now we have the annotations and are ready to calculate the ‘GeneScores’. The add_melded_assay is the DataStore method that will be used for this purpose. The add_melded_assay method is actually designed to map any arbitrary genomic coordinate information (not just gene annotations) to the ATAC peaks. Here, we use the term ‘melding’ for the process wherein for a given loci the values from all the overlapping peaks are merged/melded into that feature. The peaks values are TF-IDF normalized before melding.
Now we explain the parameters that are usually passed to the add_melded_assay.
from_assay: Name of the assay to be acted on. You can generally skip if you have only one assay. We use this parameter here only for demonstration pusposeexternal_bed_fn: This is the annotation file. Here we pass the annotation for human GRCh37/hg19 assembly based GENCODE v38 annotations.peak_col: This is the column inds.ATAC.featstable that contains the peak coordinates. In this case it is ‘ids’, but could be anyt other column. Please note that the coordinate information in this column should be in format ‘chr:start-end’ (please note the positions of colon and hyphen)renormalization: By overriding the default value of True to False here, we turned off ‘renormalization’ step. The renormalization step makes sure that all the feature values for each cells in the melded assay sum up to the same value. Here we turned this off because we will have ‘GeneScores’ as an ‘RNAassay’ which uses library size normalization that has the same effect as ‘renormalization’.assay_label: The label/name of the melded/output assay. Because we are using gene bodies as our input, ‘GeneScores’ is sensible choice.assay_type: Here we set the type of assay as ‘RNA’ which means that the new melded assay will be treated as if it was an scRNA-Seq assay. Alternatively, we could have set it as a generic ‘Assay’
ds.add_melded_assay(
from_assay='ATAC',
external_bed_fn='scarf_datasets/annotations/human_GRCh37_gencode_v38_gene_body.bed.gz',
peaks_col='ids',
renormalization=False,
assay_label='GeneScores',
assay_type='RNA'
)
We can now print out the DataStore and see that the ‘GeneScores’ assay has indeed been added. The ‘add_melded_assay’ also printed a useful bit of information that almost half of features(genes bodies) did not overlap with even a single peak. The melded assay anyway contains all the genes but sets these ‘empty’ genes as invalid.
ds
Let’s now visualize some ‘GeneScores’ for some of the known marker genes for PBMCs on the UMAP plot calculated on the ATAC assay
ds.plot_layout(
layout_key='ATAC_UMAP', from_assay='GeneScores',
color_by=['CD3D', 'MS4A1', 'LEF1', 'NKG7', 'TREM1', 'LYZ'],
clip_fraction=0.01,
n_columns=3,
width=3,
height=3,
point_size=5,
scatter_kwargs={'lw': 0.01},
)
We stop here in this vignette. But will soon add other vignettes that show how we can other kinds of melded assays like motifs, enhancers, etc. We will also explore how we can use the ‘GeneScores’ to integrate scATAC-Seq datasets with scRNA-Seq datasets from same population of cells (but not the exact same set of cells)
That is all for this vignette.