Workflow for scATAC-Seq data¶
[1]:
%load_ext autotime
%config InlineBackend.figure_format = 'retina'
import scarf
scarf.__version__
[1]:
'0.8.5'
time: 976 ms (started: 2021-08-22 18:35:04 +00:00)
1) Fetch and convert data¶
[2]:
scarf.fetch_dataset('tenx_10K_pbmc_atacseq', save_path='scarf_datasets')
reader = scarf.CrH5Reader('scarf_datasets/tenx_10K_pbmc_atacseq/data.h5', 'atac')
reader.assayFeats
INFO: Download started...
INFO: Download finished! File saved here: /home/docs/checkouts/readthedocs.org/user_builds/scarf/checkouts/0.8.5/docs/source/vignettes/scarf_datasets/tenx_10K_pbmc_atacseq/data.h5
2021-08-22 18:35:18 URL:https://storage.googleapis.com/cos-osf-prod-files-de-1/f6ce35654605c1e034e29fd1bc9fedd4feb947b937d7eec8a1ddaff4cdb19cc8?response-content-disposition=attachment%3B%20filename%3D%22data.h5%22%3B%20filename%2A%3DUTF-8%27%27data.h5&GoogleAccessId=files-de-1%40cos-osf-prod.iam.gserviceaccount.com&Expires=1629657374&Signature=rTsWiJOUE0kCOI3TJGrFIk8eV9mAjjMQIODGFiixeJcGloUKcLeKD9gxp5DevtxDQtGdN9j1dtSzxWV3CQq5y8ACT2ikK5teSedPOrfLqzIHm8j1GNpAtCWKMxvGd1FqIvg1yi2WOvphPVM5QrUKXsmvUCMJsAK9%2B2y8FWss7DKPz5aqWmyGaUt4Z2lKeQfNE2wqAuxWb3honjAVmW8491nA4cy8RvJi%2FrKwCvJIL35Tyxc1FflkaqG8%2BFP8eVa3MUsH%2FQ%2B74BikF8W0QtGasSt7b9F69crIqmAC4zQ14GAUOtMgpqWoZb0IdaPDBN2r9Hge%2F5UNpzFHqc5KMvA%2F7A%3D%3D [101645338/101645338] -> "/home/docs/checkouts/readthedocs.org/user_builds/scarf/checkouts/0.8.5/docs/source/vignettes/scarf_datasets/tenx_10K_pbmc_atacseq/data.h5" [1]
[2]:
| ATAC | |
|---|---|
| type | Peaks |
| start | 0 |
| end | 90686 |
| nFeatures | 90686 |
time: 13.1 s (started: 2021-08-22 18:35:05 +00:00)
[3]:
writer = scarf.CrToZarr(reader, zarr_fn=f'scarf_datasets/tenx_10K_pbmc_atacseq/data.zarr', chunk_size=(1000, 2000))
writer.dump(batch_size=1000)
100%|██████████| 10/10 [00:16<00:00, 1.61s/it]
time: 16.4 s (started: 2021-08-22 18:35:18 +00:00)
2) Create DataStore and filter cells¶
[4]:
ds = scarf.DataStore('scarf_datasets/tenx_10K_pbmc_atacseq/data.zarr', nthreads=4)
INFO: Setting assay ATAC to assay type: ATACassay
INFO: (ATAC) Computing nCells and dropOuts
[########################################] | 100% Completed | 3.1s
INFO: (ATAC) Computing nCounts
[########################################] | 100% Completed | 2.8s
INFO: (ATAC) Computing nFeatures
[########################################] | 100% Completed | 3.0s
time: 9.02 s (started: 2021-08-22 18:35:35 +00:00)
[5]:
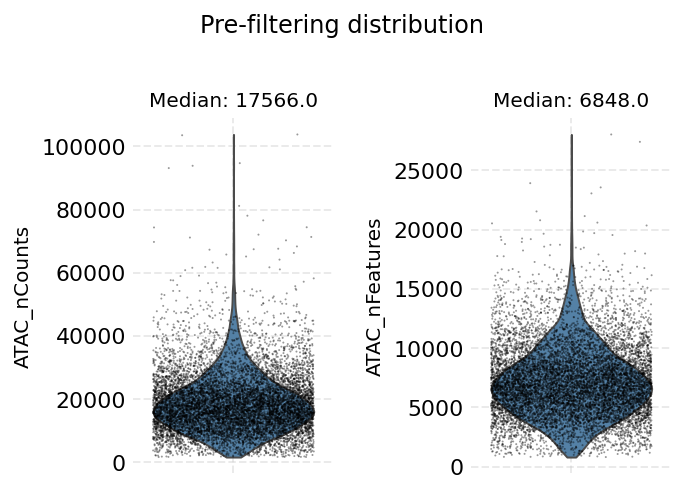
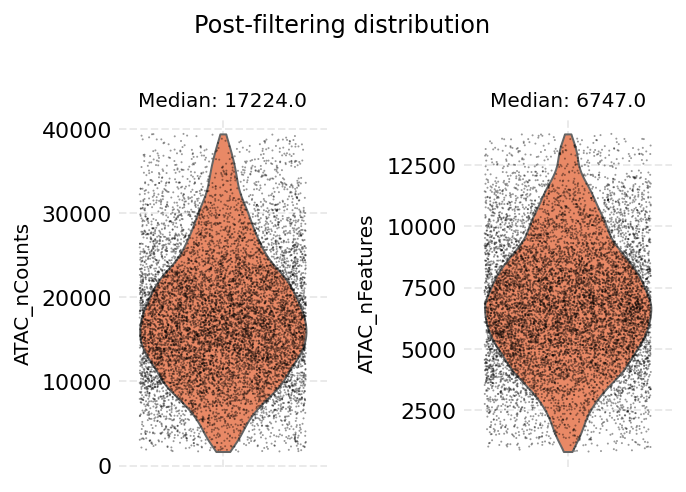
ds.auto_filter_cells()
INFO: 296 cells flagged for filtering out using attribute ATAC_nCounts
INFO: 260 cells flagged for filtering out using attribute ATAC_nFeatures
time: 4.63 s (started: 2021-08-22 18:35:44 +00:00)
3) Feature selection¶
For scATAC-Seq data, the features are ranked by their TF-IDF normalized values, summed across all cells. The top n features are marked as prevalent_peaks and are used for downstream steps.
[6]:
ds.mark_prevalent_peaks(top_n=20000)
INFO: (ATAC) Calculating peak prevalence across cells
[########################################] | 100% Completed | 7.4s
time: 7.56 s (started: 2021-08-22 18:35:48 +00:00)
4) KNN graph creation¶
For scATAC-Seq datasets, Scarf uses TF-IDF normalization. The normalization is automatically performed during the graph building step. The selected features, marked as prevalent_peaks in feature metadata, are used for graph creation. For the dimension reduction step, LSI (latent semantic indexing) is used rather than PCA. The rest of the steps are same as for scRNA-Seq data.
[7]:
ds.make_graph(feat_key='prevalent_peaks', k=11, dims=21, n_centroids=1000)
INFO: No value provided for parameter `log_transform`. Will use default value: True
INFO: No value provided for parameter `renormalize_subset`. Will use default value: True
INFO: No value provided for parameter `pca_cell_key`. Will use default value: I
INFO: Using LSI for dimension reduction
INFO: No value provided for parameter `ann_metric`. Will use default value: l2
INFO: No value provided for parameter `ann_efc`. Will use default value: min(100, max(k * 3, 50))
INFO: No value provided for parameter `ann_ef`. Will use default value: min(100, max(k * 3, 50))
INFO: No value provided for parameter `ann_m`. Will use default value: 48
INFO: No value provided for parameter `rand_state`. Will use default value: 4466
INFO: No value provided for parameter `local_connectivity`. Will use default value: 1.0
INFO: No value provided for parameter `bandwidth`. Will use default value: 1.5
Writing data to normed__I__prevalent_peaks/data: 100%|██████████| 10/10 [00:13<00:00, 1.38s/it]
/home/docs/checkouts/readthedocs.org/user_builds/scarf/envs/0.8.5/lib/python3.8/site-packages/gensim/similarities/__init__.py:15: UserWarning: The gensim.similarities.levenshtein submodule is disabled, because the optional Levenshtein package <https://pypi.org/project/python-Levenshtein/> is unavailable. Install Levenhstein (e.g. `pip install python-Levenshtein`) to suppress this warning.
warnings.warn(msg)
Fitting LSI model: 100%|██████████| 10/10 [01:04<00:00, 6.46s/it]
Fitting ANN: 100%|██████████| 10/10 [00:04<00:00, 2.44it/s]
Fitting kmeans: 100%|██████████| 10/10 [00:05<00:00, 1.86it/s]
Estimating seed partitions: 100%|██████████| 10/10 [00:03<00:00, 2.57it/s]
INFO: Saving loadings to ATAC/normed__I__prevalent_peaks/reduction__lsi__21__I
INFO: Saving ANN index to ATAC/normed__I__prevalent_peaks/reduction__lsi__21__I/ann__l2__50__50__48__4466
INFO: Saving kmeans clusters to ATAC/normed__I__prevalent_peaks/reduction__lsi__21__I/kmeans__1000__4466
Saving KNN graph: 100%|██████████| 10/10 [00:03<00:00, 2.59it/s]
INFO: ANN recall: 99.91%
Smoothening KNN distances: 100%|██████████| 2/2 [00:02<00:00, 1.14s/it]
time: 1min 58s (started: 2021-08-22 18:35:56 +00:00)
5) UMAP reduction and clustering¶
Non-linear dimension reduction using UMAP and tSNE are performed in the same way as for scRNA-Seq data. Similarly the clustering step is also performed in the same way as for scRNA-Seq data.
[8]:
ds.run_umap(fit_n_epochs=250, min_dist=0.5, parallel=True)
completed 0 / 250 epochs
completed 25 / 250 epochs
completed 50 / 250 epochs
completed 75 / 250 epochs
completed 100 / 250 epochs
completed 125 / 250 epochs
completed 150 / 250 epochs
completed 175 / 250 epochs
completed 200 / 250 epochs
completed 225 / 250 epochs
completed 0 / 100 epochs
completed 10 / 100 epochs
completed 20 / 100 epochs
completed 30 / 100 epochs
completed 40 / 100 epochs
completed 50 / 100 epochs
completed 60 / 100 epochs
completed 70 / 100 epochs
completed 80 / 100 epochs
completed 90 / 100 epochs
time: 17.9 s (started: 2021-08-22 18:37:55 +00:00)
[9]:
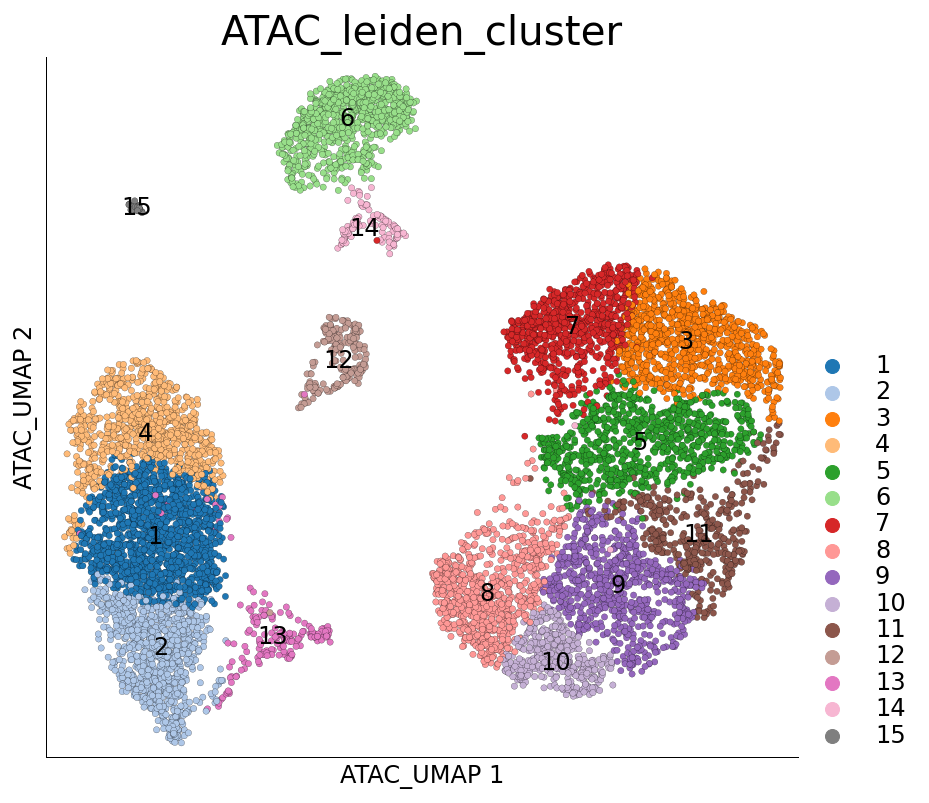
ds.run_leiden_clustering(resolution=1)
time: 214 ms (started: 2021-08-22 18:38:13 +00:00)
[10]:
ds.plot_layout(layout_key='ATAC_UMAP', color_by='ATAC_leiden_cluster')
time: 686 ms (started: 2021-08-22 18:38:13 +00:00)
6) Calculating gene scores¶
This feature is coming soon..
[11]:
ds.ATAC.feats.head()
[11]:
| I | ids | names | I__prevalent_peaks | dropOuts | nCells | stats_I_prevalence | |
|---|---|---|---|---|---|---|---|
| 0 | True | chr1:565163-565491 | chr1:565163-565491 | False | 9619 | 49 | 0.123722 |
| 1 | True | chr1:569190-569620 | chr1:569190-569620 | False | 9545 | 123 | 0.299672 |
| 2 | True | chr1:713551-714783 | chr1:713551-714783 | True | 6403 | 3265 | 2.214712 |
| 3 | True | chr1:752418-753020 | chr1:752418-753020 | False | 9102 | 566 | 0.557693 |
| 4 | True | chr1:762249-763345 | chr1:762249-763345 | True | 7433 | 2235 | 1.673136 |
time: 26.3 ms (started: 2021-08-22 18:38:13 +00:00)
That is all for this vignette.